




Page suivante: 4.2.2 Détermination des topologies des Niveau précédent: 4.2 Détermination de structures de Page précédente: 4.2 Détermination de structures de
Page suivante: 4.2.2 Détermination des topologies des
Niveau précédent: 4.2 Détermination de structures de
Page précédente: 4.2 Détermination de structures de
La Résonance Magnétique Nucléaire (RMN) est une méthode d'investigation puissante pour le biochimiste. Une expérience standard de RMN produit un spectre reflétant les interactions entre atomes présentant un moment magnétique ou spin. Dans les protéines, les moments magnétiques de certains atomes étant très sensibles à l'environnement chimique immédiat, le spectre fourni par une expérience peut donner des informations sur la structure moléculaire. Dans le spectre, chaque pic est représentatif d'une interaction entre atomes. Ces interactions peuvent mettre en jeu des atomes d'un seul acide aminé (interactions intra-résidu) ou des atomes d'acides aminés distincts (interactions inter-résidus). L'ensemble des pics (système de spins) généré par un acide aminé dans un spectre intra-résidu permet de caractériser partiellement le type de cet acide aminé.
Les interactions entre paires d'atomes sont mises en évidence dans un spectre à deux dimensions. La RMN à trois ou quatre dimensions, plus informative, permet d'obtenir des spectres mettant en évidence des interactions entre triplets ou quadruplets d'atomes. L'utilisation de la RMN dans le cadre de l'élucidation de structures complexes de molécules biologiques, les protéines par exemple, se décompose généralement en 4 étapes :
Le système AUTOASSIGN [ Zim 93] s'appuie sur l'analyse de deux types de spectres RMN à trois dimensions pour résoudre l'étape d'affectation (cf. Figure 7). Le premier spectre est spécifique des relations intra-résidu. Il permet d'affecter à chaque système de spins l'ensemble des acides aminés de la séquence dont ce système de spins est caractéristique. L'affectation précédente étant ambiguë et un même type d'acide aminé pouvant avoir plusieurs occurrences dans la séquence, elle ne suffit pas à associer un système de spins à un acide aminé particulier de la séquence. Il est heureusement possible d'utiliser un spectre inter-résidus qui fournira, pour un système de spins donné, des informations sur les systèmes de spins adjacents (précédent et suivant).
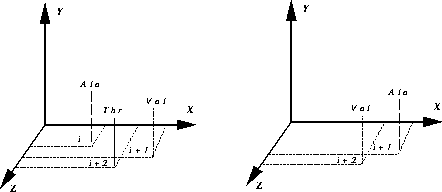
Figure 7: Schématisation des deux spectres RMN utilisés pour l'affection
des systèmes de spins aux acides aminés. La séquence est composée de 3
acides aminés dont chacun possède un système de spins caractéristique. À
gauche, le spectre spécifique des interactions intra-résidus. On a pu
affecter à chaque système de spins un type d'acide aminé (Ala, Thr,
Val). Dans la séquence, Ala est en position i, Val en position i+1
et Thr en position i+2. À droite, le spectre des relations inter
acides aminés. En chaque point du plan XZ associé à un système de
spins correspond le type de résidu de l'acide aminé voisin. La relation
entre les deux spectres est une simple translation dans le plan XZ.
Le système AUTOASSIGN [ Zim 93] utilise le cadre formel des CSP pour traiter le problème d'affectation suivant : étant donnée la séquence d'acides aminés de la protéine et les spectres spécifiques des interactions intra/inter-résidus, attribuer les systèmes de spins aux acides aminés de la séquence. La représentation sous la forme d'un CSP peut s'effectuer de la façon suivante :
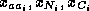 )
sont associées à chaque système de spins
)
sont associées à chaque système de spins 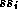 et une variable est
associée à chaque acide aminé de la séquence (
et une variable est
associée à chaque acide aminé de la séquence (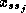 ) ; la variable
) ; la variable
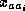 représente l'acide aminé de la séquence correspondant au
système de spins
représente l'acide aminé de la séquence correspondant au
système de spins 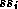 ; les variables
; les variables 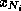 et
et 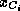 représentent les systèmes de spins des acides aminés adjacents à l'acide
aminé correspondant à
représentent les systèmes de spins des acides aminés adjacents à l'acide
aminé correspondant à 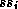 ;
;
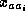 est
l'ensemble des acides aminés de la séquence ; pour les variables
est
l'ensemble des acides aminés de la séquence ; pour les variables 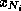 et
et 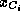 le domaine initial est l'ensemble des systèmes de spins
autres que
le domaine initial est l'ensemble des systèmes de spins
autres que 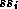 ; le domaine initial d'une variable
; le domaine initial d'une variable 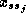 est donné
par l'ensemble des systèmes de spins ;
est donné
par l'ensemble des systèmes de spins ;
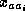 peut alors être réduit à un ensemble de valeurs qui sont les
acides aminés correspondant de la séquence ; ainsi, si un pic est
caractéristique de l'acide aminé Ala, alors le domaine de la
variable associée peut être réduit à l'ensemble des valeurs représentant
les différentes occurrences de l'acide aminé Ala dans la séquence ;
dans le deuxième spectre, des relations d'adjacence entre systèmes de
spins sont mises en évidence ; des contraintes imposent à de tels systèmes
que les acides aminés qui leur sont attribués apparaissent adjacents dans
la séquence ; par exemple, si les données (spectres) suggèrent qu'un
système de spins correspondant à un acide aminé de type A est suivi
par un système de spins correspondant à un acide aminé de type B
alors il faut que la sous-séquence AB apparaisse dans la séquence ;
enfin, il est clair que deux systèmes de spins ne peuvent être attribués à
un même acide aminé de la séquence et vice-versa.
peut alors être réduit à un ensemble de valeurs qui sont les
acides aminés correspondant de la séquence ; ainsi, si un pic est
caractéristique de l'acide aminé Ala, alors le domaine de la
variable associée peut être réduit à l'ensemble des valeurs représentant
les différentes occurrences de l'acide aminé Ala dans la séquence ;
dans le deuxième spectre, des relations d'adjacence entre systèmes de
spins sont mises en évidence ; des contraintes imposent à de tels systèmes
que les acides aminés qui leur sont attribués apparaissent adjacents dans
la séquence ; par exemple, si les données (spectres) suggèrent qu'un
système de spins correspondant à un acide aminé de type A est suivi
par un système de spins correspondant à un acide aminé de type B
alors il faut que la sous-séquence AB apparaisse dans la séquence ;
enfin, il est clair que deux systèmes de spins ne peuvent être attribués à
un même acide aminé de la séquence et vice-versa.
Les principales difficultés de ce problème ont deux sources :
Copyright(C)1995
INRA
Tous droits réservés