Logiciels
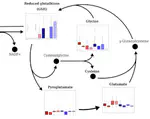


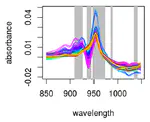
ASTERICS is a web application that aims at making complex exploratory and integration analysis workflows easily available to biologists. Edition, Exploration and Integration menus organize the interface to perform data edition, missing value imputation, normalization, data exploration with interactive plots, numerical summaries, PCA, tests, clustering, self-organizing maps, and data integration with various methods. Analyses are adapted to the most standard omics datasets. This project has been funded by Région Occitanie (Grant number 20008788).


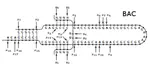
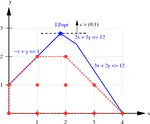
Baryonyx est un solveur de problèmes linéaires en nombres entiers et booléens. Il intègre la version généralisée de l’algorithme In the Middle de Dag Wedelin ainsi qu’une version expérimentale de gestion des fonctions objectifs quadratiques. Baryonyx fonctionne avec des exécutions répétées et simultanées sur différents processeurs afin d’améliorer la solution à chaque cycle d’exécution. Il prend en charge le format de fichiers de type LP de IBM CPLEX.





The platform works in tight collaboration with the GeT sequencing platform for the management and the analysis of data produced by their Roche 454 and Illumina HiSeq sequencers. NG6 is an extensible sequencing provider oriented LIMS. It includes read quality control and first level analysis processes which ease the data validation made jointly by the sequencing facility staff ant the end-users. It provides a secured user-friendly interface to visualize and download the raw sequences files and the analysis results.