Stage de Master 2 : Réduction de dimension pour l’analyse de données omiques spatiales
Pour toute candidature, envoyez votre CV ainsi qu’une lettre de motivation à Benjamin Charlier et Paul Esclande.
Acquisition spatialisée en biologie moléculaire (omiques spatiales)
Depuis une dizaine d’années, les technologies d’acquisition en omique spatiale permettent de localiser précisément les lectures au sein des tissus, souvent avec une résolution infracellulaire. Elles offrent ainsi la possibilité d’analyser l’état moléculaire d’une cellule dans son contexte spatial. Cette capacité à relier organisation tissulaire et fonctions biologiques ouvre un accès sans précédent à l’étude des communications intercellulaires au sein des tissus.
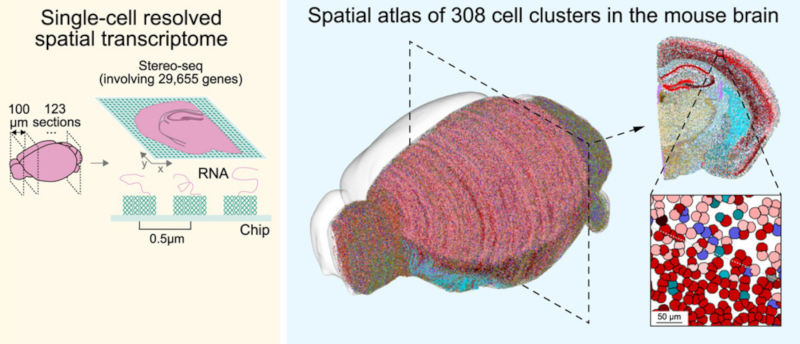
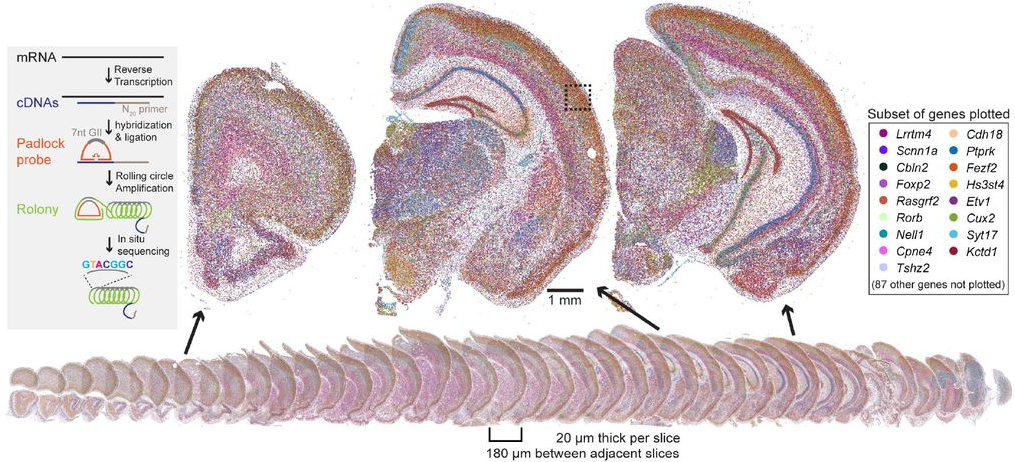
Les données constituent aujourd’hui un axe stratégique majeur pour les recherches en biologie moléculaire [0]. Toutefois, les avancées expérimentales progressent plus rapidement que les méthodes d’analyse disponibles, ce qui limite actuellement leur exploitation optimale. Les difficultés d’analyse proviennent notamment de leur très grande dimensionnalité (centaines de millions de lectures, plusieurs centaines de gènes détectables, conduisant à des fichiers de plusieurs téraoctets), ainsi que de l’absence de larges bases de référence, conséquence directe du coût encore élevé de ces acquisitions.
Méthodes de réduction de dimension
Le but du stage est d’explorer les méthodes de réduction de dimension pour les données de transcriptomique spatiale. Deux pistes principales seront approfondies :
Rééchantillonnage spatial optimisé : Une première approche pour traiter ces acquisitions consiste à adopter une stratégie multiéchelle permettant d’ajuster la résolution des données brutes en fonction des objectifs biologiques étudiés et des moyens de calcul disponibles. Il s’agit en particulier d’utiliser une approximation optimale au sens des varifolds, visant à réduire le nombre de lectures tout en préservant les particularités de chaque région [4].
Sélection de gènes : Il s’agit de choisir un sous-ensemble de gènes maximisant un critère pertinent pour répondre à une question biologique d’intérêt. Parmi les approches déjà décrites dans la littérature, on trouve notamment des méthodes basées sur la variance spatiale, qui identifient des gènes dont l’expression varie selon la localisation [5], ainsi que des approches fondées sur l’information mutuelle, favorisant les gènes dont l’expression présente une structure marquée le long de frontières bien définies [4]. Enfin, des méthodes de réduction de dimension ont également été proposées afin de faciliter l’application d’algorithmes de clustering [3].
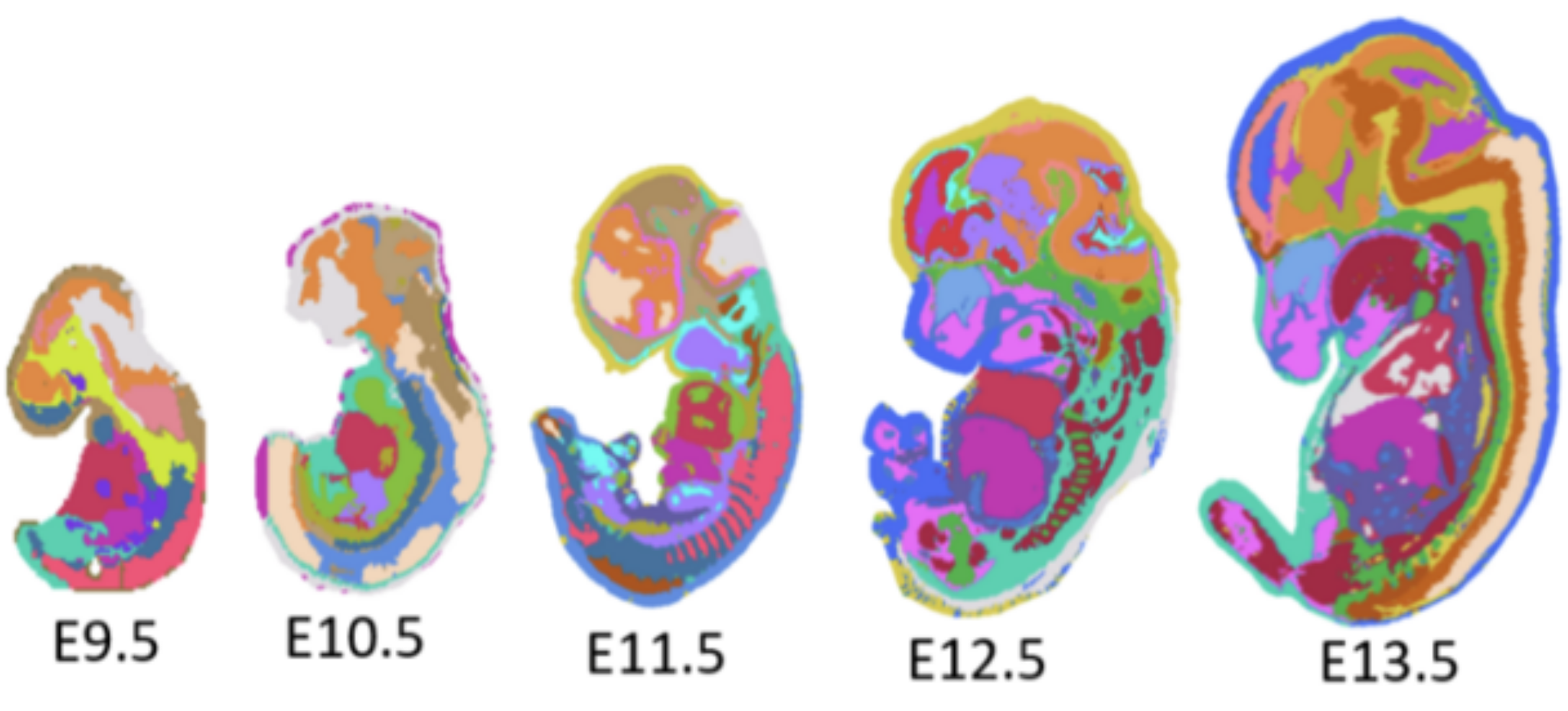
Déroulé du stage
L’objectit principal du stage sera de concevoir, développer et implémenter une méthode de selection de gènes passant à l’échelle des données réelles. Les données utilisées pour mettre en place les méthodes proviendront de bases de données publiques. Des collaborations avec une équipe au Center for Imaging Science (Johns Hopkins, Baltimore) et Marseille Medical Genetics enrichiront les applications.
A cette fin, le stage pourra envisager les récentes avancées en apprentissage profond et pourra s’articuler autour des étapes suivantes. L’étudiant.e réalisera dans un premier temps une revue de l’état de l’art des méthodes réduction de dimension et d’analyse des données de transcriptomique spatiale. En parallèle, une phase d’appropriation des outis développées par l’équipe encadrante [3,4] sera menée afin de permettre une montée en compétence rapide et une utilisation efficace des environnements de travail.
Profil recherché
Le ou la candidat·e devra être issue d’une formation de niveau master 2 en sciences ou d’un cycle ingénieur. Le sujet étant par nature pluridisciplinaire, des compétences en mathématiques appliquées et/ou en machine learning, et/ou en modélisation statistique, et/ou en bioinformatique seront mobilisées. Cette personne devra également être capable de programmer en Python et une curiosité pour les applications en biologie sera appréciée.
Cadre de travail
Le stage, d’une durée de 5 à 6 mois et rémunéré, se déroulera au sein de l’unité MIAT (Mathématiques et Informatique Appliquées de Toulouse) de l’INRAE, située à Toulouse (24, chemin de Borde Rouge, 31320 Auzeville-Tolosane).
Le Centre Inrae est facilement accessible en transports en commun (ou en vélo), dispose d’un restaurant d’entreprise, et d’une association qui propose de nombreuses activités sportives et autres.
Bien que ne disposant pas de financament dédié, le stage pourra éventuellement déboucher sur une thèse en candidatant aux concours de l’école doctorale.
Encadrement et contact
Le stage sera encadré par Benjamin Charlier (INRAE, MIAT) et Paul Escande (CNRS, IMT).
Pour toute demande d’information ou candidature, contacter les encadrants.
Références
[0] Marx, V. Method of the Year: spatially resolved transcriptomics. Nature Methods, 18, 9–14 (2021). https://doi.org/10.1038/s41592-020-01033-y
[1] Covert, I., Gala, R., Wang, T., et al. Predictive and robust gene selection for spatial transcriptomics. Nature Communications, 14, 2091 (2023). https://doi.org/10.1038/s41467-023-37392-1
[2] Khan, M., Arslanturk, S., Draghici, S. A comprehensive review of spatial transcriptomics data alignment and integration. Nucleic Acids Research, 53(12) (2025). https://doi.org/10.1093/nar/gkaf536
[3] Assali, I., Escande, P., Villoutreix, P. jsPCA: fast, scalable, and interpretable identification of spatial domains and variable genes across multi-slice and multi-sample spatial transcriptomics data. (Prépublication / référence de journal non indiquée)
[4] Stouffer, K. M., Trouvé, A., Younes, L., et al. Cross-modality mapping using image varifolds to align tissue-scale atlases to molecular-scale measures with application to 2D brain sections. Nature Communications, 15, 3530 (2024). https://doi.org/10.1038/s41467-024-47883-4
[5] Yuan, X., Ma, Y., Gao, R., et al. HEARTSVG: a fast and accurate method for identifying spatially variable genes in large-scale spatial transcriptomics.
Nature Communications, 15, 5700 (2024). https://doi.org/10.1038/s41467-024-49846-1
[6] Han, L., Liu, Z., Jing, Z., et al. Single-cell spatial transcriptomic atlas of the whole mouse brain. Cell, 113(13), 2141–2160.e9 (2025). (DOI non indiqué dans la source)
[7] Chen, X., Fischer, S., Rue, M. C. P., et al. Whole-cortex in situ sequencing reveals input-dependent area identity. Nature, 647, 203–212 (2025). https://doi.org/10.1038/s41586-024-07221-6